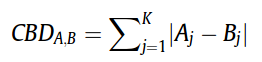-
General Information -
How to Use: Example with Metaraminol -
Glossary -
References -
Problem In Running Application?
General Information
A) Poly-Pharmacology/Promiscuous Drugs
Ability of compound to interact with more than one target proteins.B) Target Prediction
Target identification has become an area of intense research owing to it widespread application ranging from the identification of the biological active molecule, drug re-purposing, toxicity prediction to the identification of promiscuous drugs or compounds (Poly-Pharmacology). Interactions of the drug molecule with unintended side targets (off-targets) often leads to adverse drug reactions and shown to be one of the most common reason for the failure of drug molecule in the clinical trialTo evalute the Poly-pharmacology profile of given compound, range of computational target identification methods have been developed which can be broadly divided into structure and ligand based methods. Most straightforward use of structure based method, involve the concept of inverse docking: whereby single molecule docked against panel of target protein structures followed by sorting/ranking of the proteins depending upon binding free energy of ligand. In our Polypharmacology browser we are using ligand based approached which is briefly explained below.
B 1) Ligand Based Target Prediction (PPB Polypharmacology Browser)
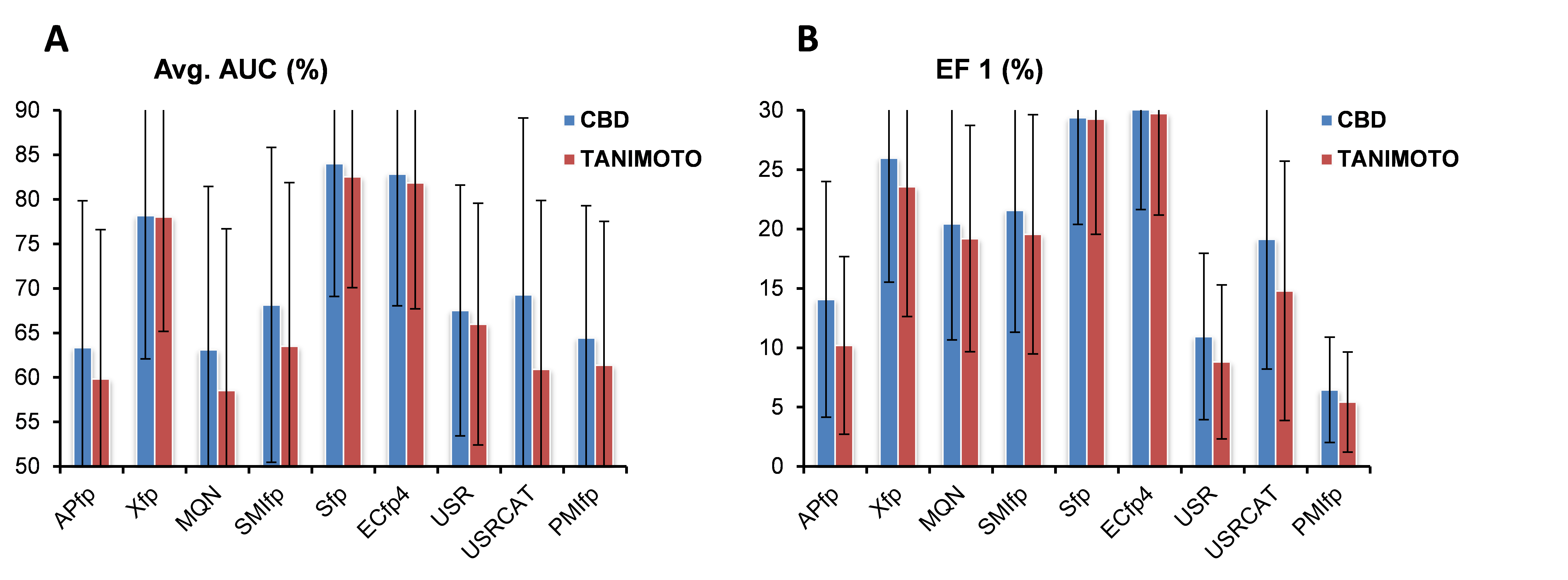
In ligand based approached each of the target protein is represented by set of known active ligands and targets are ranked depending upon similarity of molecule in question to the representative ligands of target proteins. Our Polypharmacology browser searches through more than 1463 targets proteins, each of which is represented by at least 20 bioactive molecules as reported in ChEMBL database. Similarity search is carried out by six different fingerprints (see below: Xfp, MQN, SMIfp, APfp, Sfp, ECfp4) and using four combinations of fingerprint (Hyp1=Xfp+SMIfp+Sfp, Hyp2=Xfp+MQN+SMIfp, Hyp3=Xfp+SMIfp+Sfp+ECfp4 and Hyp4=Xfp+MQN+SMIfp+Sfp+ECfp4).2D-Molecular fingerprints are well known for their use in lgand based virtual screening. Enrichment studies for recall of know-bioactive compounds of 40 target proteins from corresponding decoys reported in Directory of Useful Decoys (DUD) database shows that 2D fingerprints performed as well as or in some of the cases even better than 3D methods (shown below). Considering this fact one would expect that similarity searching in target chemical space would find most relevant nearest neighbours(close analogs) of query molecule, which can be then linked to the corresponding target proteins.
For each of the fingerprint (mentioned above), database molecules (each of the molecule is tag with its target protein) are sorted according to decreasing similarity (increasing distance) to query molecule. Following, list of top 20 targets for each fingerprint space were retrived. Lists of targets were then merge and final result is represented as shown below.
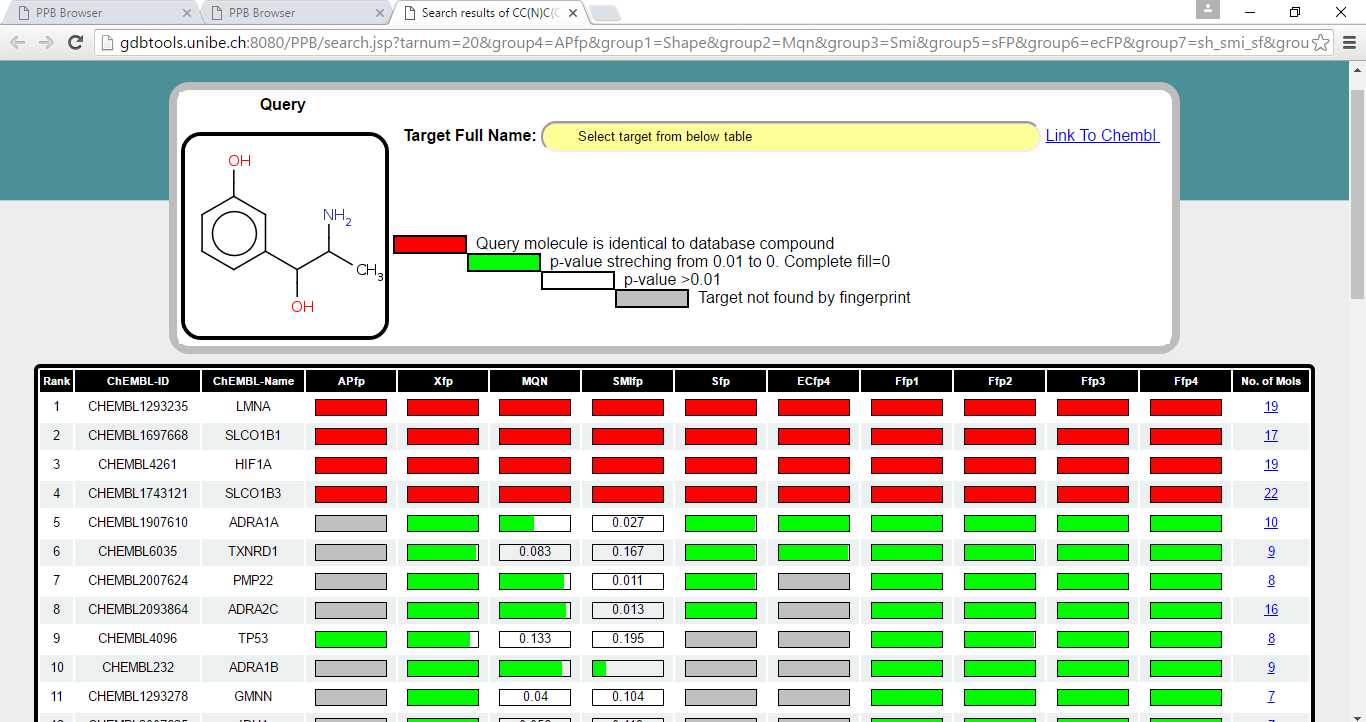
For each of the target, PValue is calculated using best match compound of "target protein" to query molecule. Green bar represent a probability streching from PValue of 0.01 to 0. When PValue is 0 bar is completly filled with green color and when PValue is more than 0.01 the bar is empty and indicated with PValue. When the target is not found by fingerprint bar is shown in grey color. When the query compound is identical to compound of target protein, bar is shown in red color.
B 2) Calculation of p-value
p-value is calculated as follows: a) for given target protein random distribution was constructed by computing the distances for up to 1M random pairs of compounds formed between known bioactive ligands of target and randomly selected compounds from ZINC database. b) random distance distribution was fitted using negative binomial distribution function available in R package. c) cumulative density plot (p-value) was generated, which gives the p-value as function of city block distance. d) given distance of query molecule to most closest nearest neighbor of given target, the p-value can be easily calculated from the cumulative density plot generated in previous step.C) Fingerprint
A vector composed of several numerical descriptors of molecular structure and properties.C 1) Topological pharmacophore (Xfp) and Atom-pair fingerprint (APfp)
Xfp pharmacophore fingerprint recently proposed from our group begins with, assignment of atoms of molecule to four different groups defined by generalized pharmacophoric atom types (see figure below), namely Hydrophobic (H), Hydrogen Bond Acceptor (HBA), Hydrogen Bond Donor (HBD) and Sp2 atoms. Following classification of atoms, Xfp counts occurrence of “atom pairs” (formed by five different groups namely H-H, HBA-HBA, HBD-HBD, HBA-HBD and Sp2-Sp2) at topological distances ranging from 0 to 10 which results in 55 (5x11) dimentional Xfp. Simpler form of Xfp called as APfp, attained by counting of all possible atom pairs at increasing topological distance (0 to 10) without considering pharmacophoric atom types, resulted in 11 dimensional topological shape fingerprint (APfp, see figure below).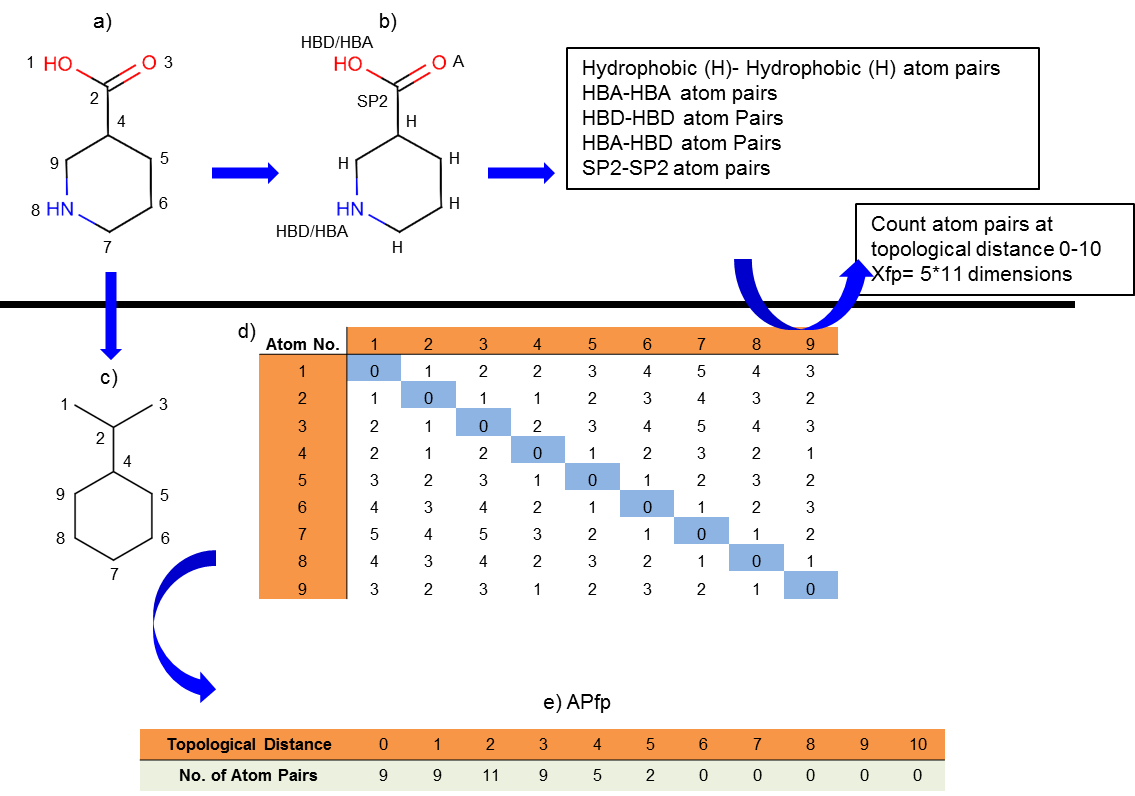
C 2) Molecular Quantum Numbers (MQN)
MQN is 42 integer value descriptors of molecular structure, which count atoms, bond types, polar groups, and topological features (shown below).
For more details please visit, MQN
C 3) SMILES Fingerprint (SMIfp)
SMIfp (SMILES fingerprint) is a scalar fingerprint counting the occurences of 34 different symbols in SMILES strings of molecules. The symbols that are counted are summarized below.
For more details please visit, SMIfp
C 4) Substructure Fingerprint (Sfp)
The Substructure fingerprint of a molecule is bit string (a sequence of "0" and "1" digits) that contains information on the structure. Substructure fingerprint (Sfp) used in the browser generated using ChemAxon JChem Package with bond length of 7 and contains 1024 bits.The Process of Fingerprint Generation in JChem:
1) Up to a given a bond number (e.g 7), all linear paths (linear patterns) consisting bonds and atoms of a structure are detected.
2) Branching points at the end of each linear pattern are also detected.
3) All cycles (cyclic patterns) are detected.
4) Using a proprietary hashing method, a given number of bits in the bit string are set for each pattern. It is possible, that the same bit is set by multiple patterns. This phenomenon is called bit collision. Few bit collisions in the fingerprint is tolerable, but too many may result in losing information in the fingerprint.
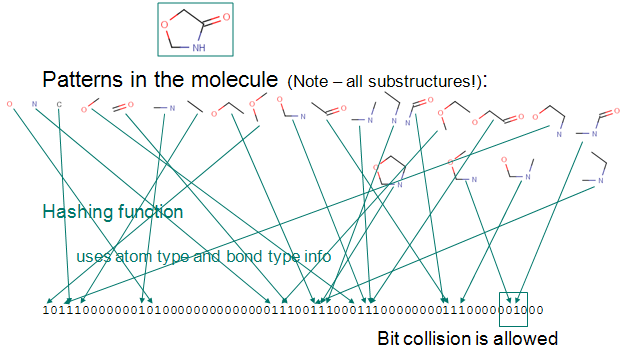
For more details visit, ChemAxon, DAYLIGHT
C 5) Extended-Connectivity Fingerprint (ECfp4)
Extended-Connectivity Fingerprints (ECFPs) are circular topological fingerprints designed for molecular characterization, similarity searching, and structure-activity modeling. They are among the most popular similarity search tools in drug discovery and they are effectively used in a wide variety of applications.Similar to Sfp, ECfp4 also encode substructure patterns from molecules on to the bit string of length 1024. Prime Difference in Sfp and ECfp4 is the way they percieve the substructure patterns from molecule. Unlike the Sfp which considered the linear path upto certain bond lenth, ECFP generate the patterns by considering the atoms into multiple circular layers up to a given diameter. In our browser we have used the ECFP with diameter of 4 (ECfp4).
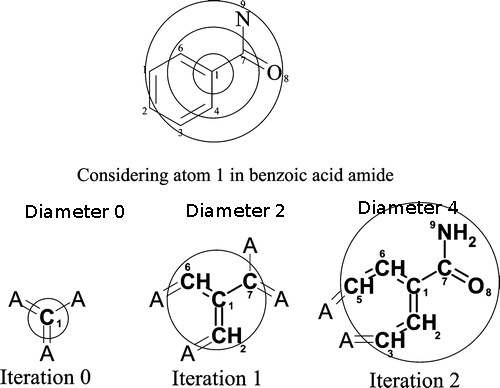
Circular patterns were generated for every atom, followed by hashing of patterns on to the bit string of length 1024. For more details please visit, ChemAxon, ECfp4
C 6) Hyp1, Hyp2, Hyp3 and Hyp4
As different fingerprints perceive same molecule in different way, consolidation of information from these fingerprints which are complementing each other’s expected to provide a new view of chemical space with the more detail and efficient encoding of small molecule.Hyp1-Hyp4 represents the four new fingerprints generated by merging of some of the parent fingerprints mentioed above.
Hyp1--> Xfp+SMIfp+Sfp
Hyp2--> Xfp+MQN+SMIfp
Hyp3--> Xfp+SMIfp+Sfp+ECfp4
Hyp4--> Xfp+MQN+SMIfp+Sfp+ECfp4
While merging, we have paid due attention to the nature of distance distribution in each of fingerprint space. As simple combination of two fingerprints (for instance Xfp and SMIfp) which have very different distance distribution mostly overshadow the effect of the one of the fingerprint having comparatively smaller distances (SMIfp). To overcome this problem we have scaled all of the dimensions of five fingerprints so as to assured the equal contribution of each fingerprint after merging.

D) (Dis) Similarity Measure
(Dis) Similarity Measure is a mathematical calculation that quantify similarity or dis-similarity between two objects (molecules). In the browser presented here, we have used City Block Distance (CBD) to calculate dis-similarity (distance) of molecules with respect to given query molecule. The list of retrieved compounds then sorted according to increasing CBD distance with respect to query.Once represented in the form of fingerprint(vector), the City block distance between two molecules(CBD A,B), A and B, with K dimensions is calculated as: